
清华大学,唯一单位发Nature正刊,全员中文署名!
分子骨架编程调控硫电化学,锂硫电池性能突破
鉴于此,来自清华大学深圳国际研究生院的周光敏副教授提出了“前介体”与分子骨架编程的概念。研究者选择2-氯嘧啶(CPyr)作为模型前介体,其可在硫反应进程中通过芳香亲核取代(SNAr)原位活化为具有介体活性的2-嘧啶硫醇锂(PyrSLi),并形成均匀覆盖电极的快速氧化还原循环。通过结合量子化学和机器学习,研究者开发了分子骨架编程策略,揭示了侧链基团的电子、几何和位点特征与介体性能之间的构效关系,实现对前介体活化速率和介导活性的调控。在196个候选分子中,2-氯-4-(三氟甲基)嘧啶(4-CF₃-CPyr)脱颖而出,使锂硫纽扣电池在800次循环中保持81.7%的容量,并在14.2 Ah级软包电池中实现了549 Wh kg⁻¹的能量密度。该策略有望拓展至更广泛的有机化学空间中功能分子的设计。相关成果以题为“Molecular skeleton programming of premediators in sulfur electrochemistry”发表在最新一期《nature》上。

SNAr反应与原位活化机制
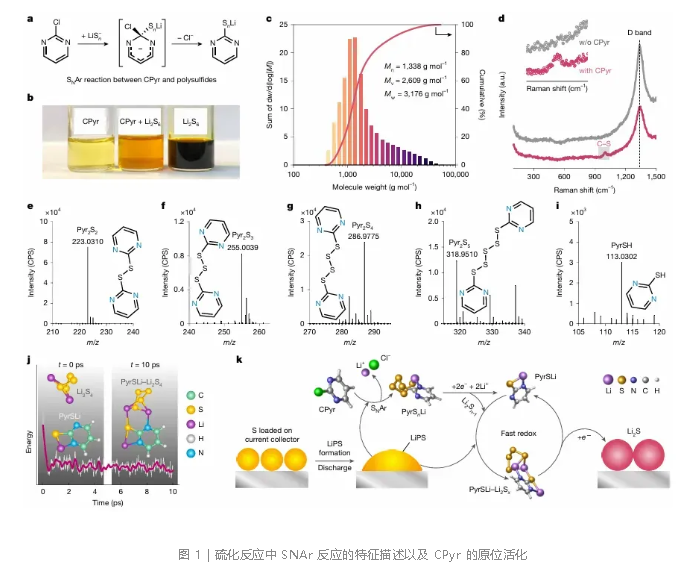
研究首先确认了CPyr与多硫化锂之间的SNAr反应。CPyr中的Cl原子作为离去基团,邻近的吸电子N原子进一步促进多硫阴离子的亲核进攻,从而将—SₙLi嫁接到嘧啶环上(图1a)。将CPyr与Li₂S₆溶液混合后,体系变为亮橙色并生成絮状物(图1b),凝胶渗透色谱显示出寡聚簇状结构(图1c),多种光谱和质谱分析确认了C—S键的形成。在实际电池中,CPyr在硫正极放电产生的多硫化物前沿被原位化学激活,生成活性吡啶硫醇盐。拉曼光谱在放电态正极检测到C—S信号(图1d),证实了电解液微环境中发生了SNAr反应。飞行时间二次离子质谱显示,CPyr体系减轻了锂负极的多硫化物腐蚀并抑制了穿梭效应。超高效液相色谱-四极杆飞行时间质谱跟踪了循环过程中的硫醇盐演变(图1e–i),揭示了以PyrS₁为核心的可逆氧化还原对,其充电态主要产物为联嘧啶二硫化物(Pyr₂S₂),放电态主要为PyrS₁。从头算分子动力学模拟表明,PyrS₁通过N—Li和S—Li键与Li₂S₄快速螯合,形成络合物后能隙降低,促进了电荷转移(图1j)。由此构筑的介导机制如下:放电时,CPyr与多硫化物反应生成活性吡啶硫醇盐;PyrS₁与多硫化物动态螯合形成低溶解度团簇,将多硫化物限域在反应前沿,并构成原位介导层,加速电荷转移。Li₂S脱离后,PyrS₁被重新释放,进入下一介导循环,从而建立起自增强的氧化还原环路(图1k)。
分子骨架设计:量子化学与机器学习融合
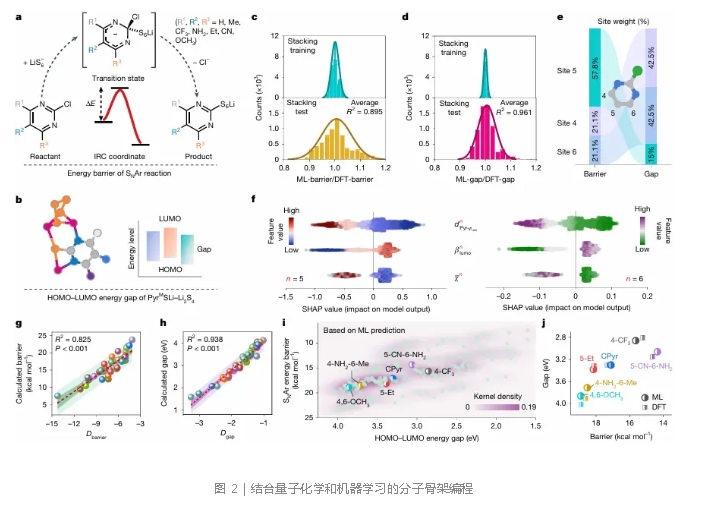
为进一步提升介导性能,作者通过分子骨架设计来调控电子特性。理想的前介体需兼具低的SNAr能垒(快速活化)和窄的PyrᴹS₁–LiPS络合物HOMO-LUMO能隙(高介导活性)。以—H、—Me、—CF₃等7种具有不同电子效应的官能团为侧链修饰单元,在嘧啶环4、5、6位点接枝,组合出196个候选分子。从40个代表性分子构成的库中,通过量子化学计算获得SNAr能垒和对应络合物的HOMO-LUMO能隙(图2a,b)。经三阶段特征工程筛选,集成学习模型对能垒和能隙的预测平均R²分别达到0.895和0.961(图2c,d)。特征重要性分析表明,5号位点对能垒预测贡献57.8%,而4、6号位点对能隙预测合计贡献85%(图2e)。静电势极小值至嘧啶环心的距离(dPyr−φmin)、beta LUMO(βlumo)和平均电负性(χ̅)被识别为关键特征(图2f)。基于此,研究者利用dPyr−φmin和χ̅构建了无量纲的能垒指数(Ibarrier)和能隙指数(Igap),并结合各位点权重定义了杂化描述符Dbarrier和Dgap,它们与理论计算值呈显著线性相关(图2g,h,R²分别为0.825和0.938)。在196个候选分子的能垒-能隙图谱中,4-CF₃-CPyr因同时具备低能垒和窄能隙而被预测为高效前介体(图2i,j)。
硫氧化还原动力学评估
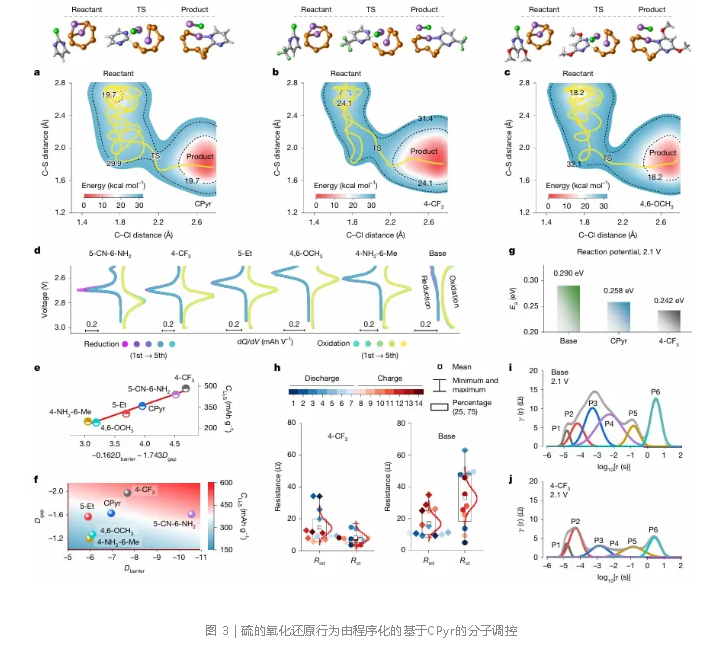
元动力学模拟直观对比了4-CF₃-CPyr、CPyr和4,6-OCH₃-CPyr与Li₂S₆反应的SNAr势能面(图3a–c),确认4-CF₃-CPyr所需活化能垒最低,活化最快。差分电容曲线显示,不同骨架的前介体均在2.7 V附近呈现可逆氧化还原对,但峰位发生偏移,表明骨架调控改变了电子结构,而活化与介导机制得以保持(图3d)。恒电位Li₂S沉积实验表明,4-CF₃-CPyr电极的Li₂S沉积容量最高(图3e,f),且该容量与活化速率和介导活性呈协同关联。变温阻抗测试求得4-CF₃-CPyr在2.1 V放电平台的活化能仅为0.242 eV,低于CPyr和空白对照(图3g)。原位电化学阻抗谱显示,其界面电阻和电荷转移电阻均显著降低,电荷转移电阻仅为空白电解液的四分之一(图3h)。弛豫时间分布分析进一步表明,4-CF₃-CPyr大幅减少了与正极电荷转移相关的极化(图3i,j)。
电池性能与有限元模拟
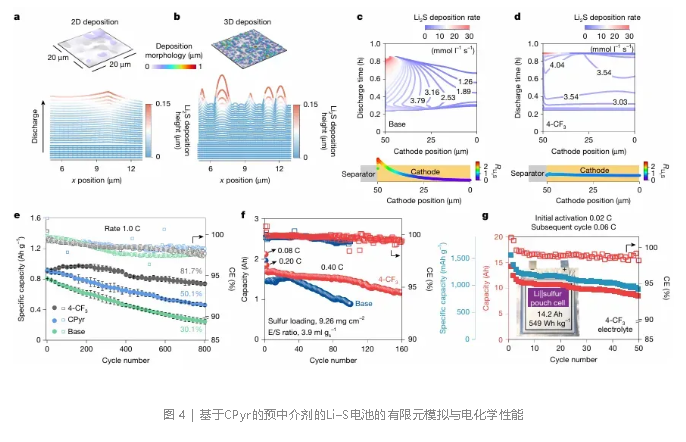
有限元模拟显示,传统正极中Li₂S倾向于在隔膜附近优先形核并呈二维横向生长,而前介体原位活化后,可在电极各处均匀引发介导循环,促使Li₂S呈三维多位点生长,提高沉积均匀性和速率(图4a–d)。纽扣电池测试中,4-CF₃-CPyr在1C倍率下循环800次后容量保持率高达81.7%(图4e)。在实际安时级软包电池中,硫载量9.26 mg cm⁻²的2.12 Ah电芯初始能量密度达342 Wh kg⁻¹,稳定循环160次;进一步将硫载量提升至28 mg cm⁻²、电解液/硫比3.4 mL g⁻¹的14.2 Ah电芯,实现了549 Wh kg⁻¹的能量密度,并可稳定循环50次(图4f,g),在同类锂硫电池中展现出标杆性能。
该工作不仅提供了一种通过原位化学活化与分子骨架编程协同提速硫转化动力学的有效前介体,更建立了一套融合量子化学与机器学习的理性分子设计范式。所揭示的构效关系与杂化描述符为调控活化速率和介导活性提供了可解释的工具,且该编程策略有望推广至有机液流电池活性材料、锂金属电池溶剂、锂-空气电池氧化还原介体以及退役正极直接修复用有机再锂化试剂等广泛的分子设计中,为能源存储系统功能分子的按需剪裁开辟了全新路径。
赞一个











更有众多热门





