
科学家们透露,前列腺素D2受体DP1的分子机制
研发家
|
2025-05-31
22
中国科学院上海药物研究所研究员徐华强和吴灿荣分析了人源前列腺素D2(PGD2)和合成激动剂BW245C的高分辨率结构,以及DP1受体无配体结合的非激活状态结构,并揭示了DP1受体激活、配体识别等分子机制,为新型DP1激动剂和拮抗剂的开发提供了坚实的结构基础,选择性更高,脱靶效果更小,预计将为各种与DP1相关的疾病提供新的治疗策略。5月26日,美国科学院发表了相关研究。
通过环氧合酶,前列腺素是通过的(COX)以这种方式产生的生物活性脂类与特定的脂类G蛋白偶联受体有关(GPCR)相互作用诱发多种生物功能,在细胞发炎、免疫反应等过程中发挥重要作用。PGD2主要在中枢系统和免疫系统中获得,在睡眠调节和炎症反应中发挥重要作用。DP1是一种典型的A类GPCR,在调节各种关键生命过程中发挥着重要作用,如睡眠、生理性炎症反应和心血管功能。然而,DP1的激活机制已经很久没有被澄清了,这限制了受体的精确药物开发。
研究小组使用冷冻电子显微镜(cryo-EM)该技术首次分析了非激活状态和激活状态下人源DP1受体的高分辨率结构。两种激活状态结构是DP1-Gs复合物结合内源激动剂PGD2,DP1-Gs复合物结合生成激动剂BW245C,以及非配体结合 DP1非激活结构。根据一系列实验和分析结果,DP1具有独特的构象特征。此外,研究团队还阐明了DP1非经典受体激活机制、配体选择性结构基础及其与G蛋白偶联的特点。
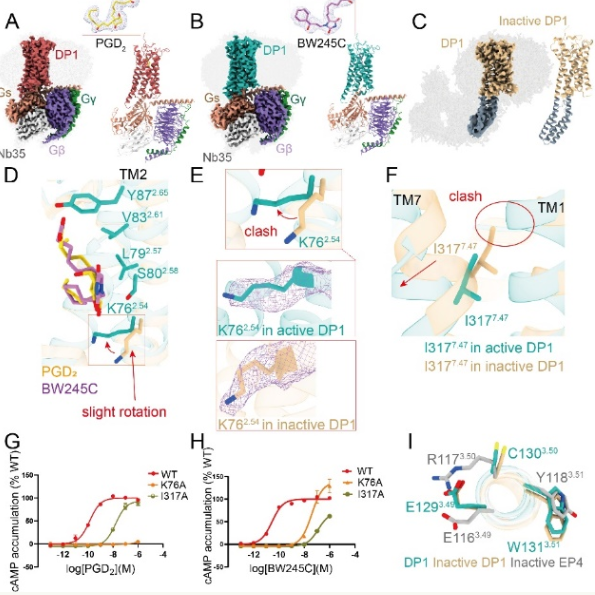
研究小组表示,该研究首次揭示了DP1受体激活和GS偶联的3D结构图和DP1受体非激活3D结构图,完善了前列腺素受体家族结构信息,为未来开发靶向DP1的高选择性药物提供了理论依据。
赞一个
22
版权及免责声明:本网站所有文章除标明原创外,均来自网络。登载本文的目的为传播行业信息,内容仅供参考,如有侵权请联系删除。文章版权归原作者及原出处所有。本网拥有对此声明的最终解释权
更多服务

推荐会议
更多 >>










最新文章
NEW
热点资讯
HOT
学术资源免费领取
加微信领取20G科研大礼包!
更有众多热门
更有众多热门




RDLINK研发家 版权所有 Copyright©2023 All rights reserved

